日前,新葡萄8883官网AMG米文慧教授及合作者对无轨道密度泛函理论进行系统综述,相关研究成果以“Orbital-Free Density Functional Theory: An attractive electronic structure method for large-scale first-principles simulations”为题,于2023年10月23日在线发表于《Chemical Reviews》。Chemical Reviews (2023年影响因子:62.1)是由美国化学学会于1924年创刊并发行至今的国际化学领域内最具权威性的学术期刊之一,致力于发表最具影响力的综述文章,以综合性、权威性以及可读性强而著称。

基于量子力学的第一性原理计算方法可以准确模拟出物质的微观状态以及其宏观性质。特别是Kohn-Sham密度泛函理论(KS-DFT),由于其兼具计算精度和效率,被广泛应用于物理、化学、材料等多个学科领域, 成为最具影响力的第一性原理计算方法。然而,传统KS-DFT由于需要对KS轨道的求解其计算量随模拟体系的增大呈超线性增长,导致其仅适用于模拟原子数目一般限于千原子的系统。大多数真实材料(如合金、玻璃、复合材料等)的模拟往往需要对数万甚至百万原子系统开展研究。无轨道密度泛函理论(OF-DFT), 正如其名所示,由于规避了轨道(波函数)的使用,大大简化了计算的复杂度,具有计算量线性标度的优势。因此,相比传统KS-DFT,OF-DFT使得能够探索更大的系统规模和更长的模拟时间尺度,从而可以探索新的化学现象和新材料,成为开展百万原子量级大尺度材料模拟的理想候选。同时,由于没有轨道信息,如何只根据电子密度构建准确动能密度泛函等泛函形式成为该领域的最大挑战。
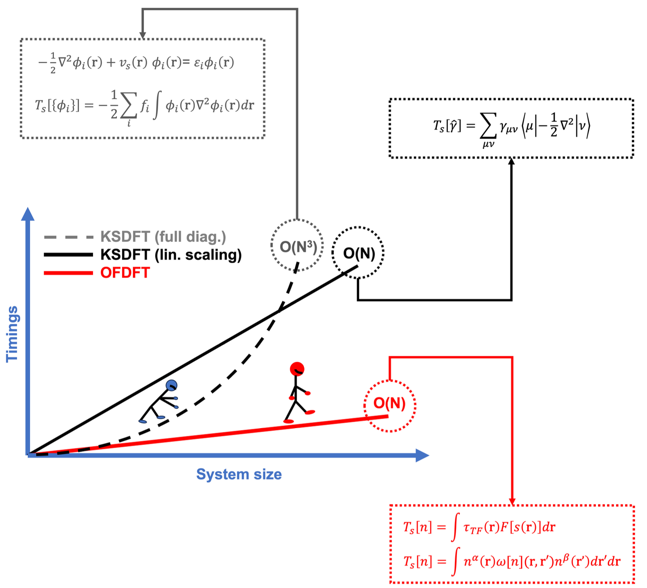
米文慧教授长期致力于OF-DFT的研究,做出系列标志性成果。该综述对OF-DFT基础理论背景,核心动能泛函的发展,数值实现过程中的数值技巧、算法及主流计算软件近一个世纪的发展进行全面系统的综述。最后,以一些应用示例作为结尾,展示了OF-DFT在材料科学、化学和物理中的强大应用潜力,为相关领域的研究工作者提供了有益的参考。
该文章第一作者兼通讯作者为物质模拟方法与软件教育部重点实验室米文慧教授,第一单位为新葡萄8883官网AMG,共同通讯作者为南京理工大学罗凯副教授,美国佛罗里达大学 Samuel B. Trickey教授,以及罗格斯大学Michele Pavanello教授。
该工作得到了新葡萄8883官网AMG物质模拟方法与软件教育部重点实验室、超硬材料国家重点实验室、未来科学国际联合实验室高压科学与技术团队的支持,得到了国家自然科学基金项目等基金的资助。
论文链接:https://pubs.acs.org/doi/10.1021/acs.chemrev.2c00758

